
Written by
Sponsored by

Proteins – the biological “workhorses” of cells – are integral to life, providing regulatory, signaling, structural, metabolic, transport and immune functions, among other roles.
A term that was first coined in 1994 by Professor Marc Wilkins, proteome refers to the entire collection of proteins that can be expressed in a cell, tissue or organism.
Proteomics refers to the study of the proteome. Examples of proteins that might be studied include:
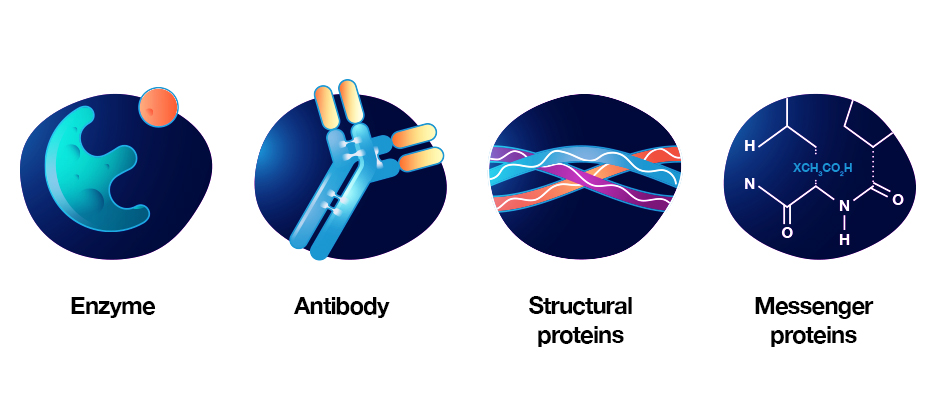
Enzymes: Enzymes are key for the thousands of chemical reactions that take place both within and between cells. They are also important for synthesizing new molecules, such as the transcription and translation of DNA, to RNA to protein.
Antibodies: Key players in the function of the immune system. Antibodies bind to foreign substances, flagging them for neutralization or destruction by cells of the immune system.
Structural proteins: Offer support and stability to cells and enable movement in certain types of cells.
Messenger proteins: Important for communication within and between cells. Messenger proteins help to transmit signals that coordinate biological processes.
Click on the individual protein examples to learn more.
Proteomics research can offer a comprehensive understanding of the molecular processes that underpin different biological states, such as healthy or diseased processes, in different cells, tissues or organisms.
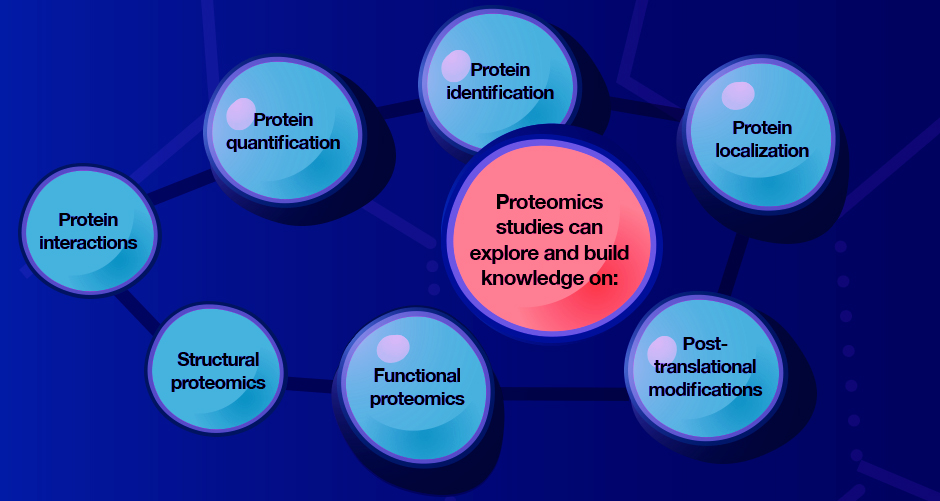
Proteomics research methods are often categorized as either “top down” or “bottom up”, which reflects whether the sample is separated prior to the peptides being analyzed. Choosing which approach to take typically depends on the research query.
Several different analytical techniques can be adopted in proteomics research, broadly divided into two categories: low-throughput and high-throughput.
Low-throughput
Antibody-based methods
Enzyme-linked immunosorbent assay (ELISA) and western blotting utilize antibodies targeted to specific proteins – or epitopes – to identify and quantify proteins in a sample.
Gel-based methods
A “mature” method for screening protein expression at a large-scale.
2D gel electrophoresis separates proteins according to their isoelectric point, and SDS-PAGE separates them further still, according to their molecular mass.
Chromatography-based methods
Used to separate and purify proteins from complex biological mixtures. Different types of chromatography can be used to separate proteins based on characteristics such as:
Chromatography is often used to prepare proteins for downstream high-throughput methods.
High-throughput
Microarrays
Apply small quantities of sample to a chip. Antibodies can be immobilized to the chip, capturing target proteins from a sample. Divided into different categories:
Analytical – used to measure levels of protein expression and their binding affinities.
Functional – used to characterize protein functions, e.g., interactions and enzyme-substrate turnover.
Reverse-phase – proteins from sample of interest are bound to the chip, which is then treated with specific antibodies to target these proteins.
The most comprehensive approach for quantifying and profiling proteins, their interactions and any modifications.
Many variations of workflows are adopted, but the general principle is as follows:
The level of sophistication and sensitivity of high-throughput techniques underpins many research advancements made in proteomics over recent years, and the field shows no signs of slowing down.
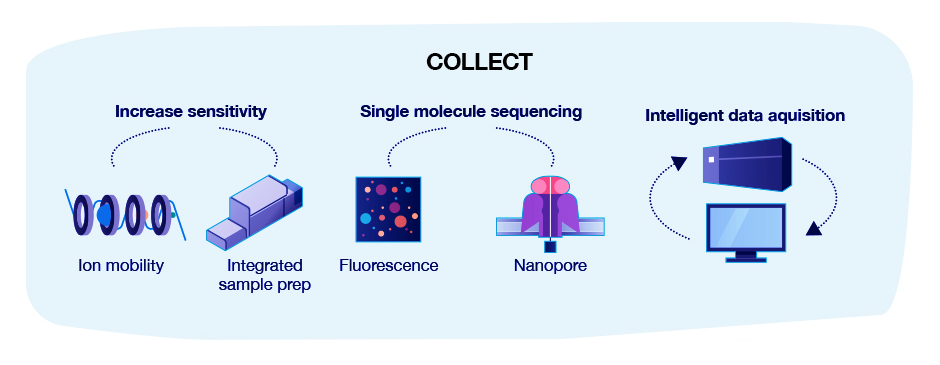
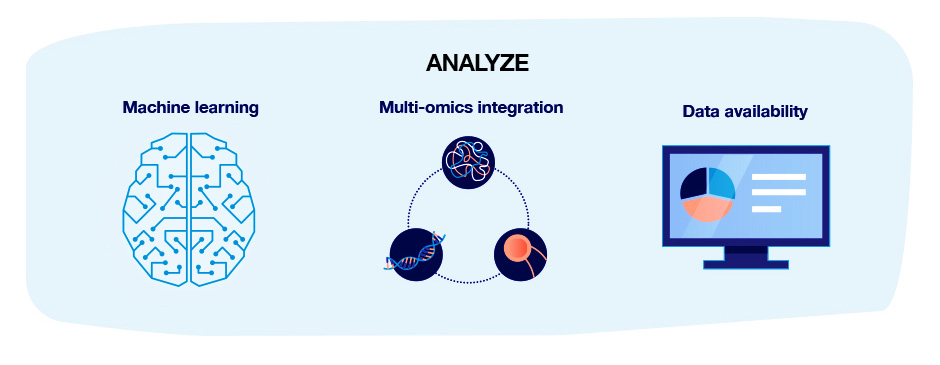
Advances in technologies that are likely to impact the utility of proteomics data can be categorized as advances in sample quality, preparation, data collection and data analysis:
The applications of proteomics research are numerous and continue to grow. Click on the key examples to learn more.
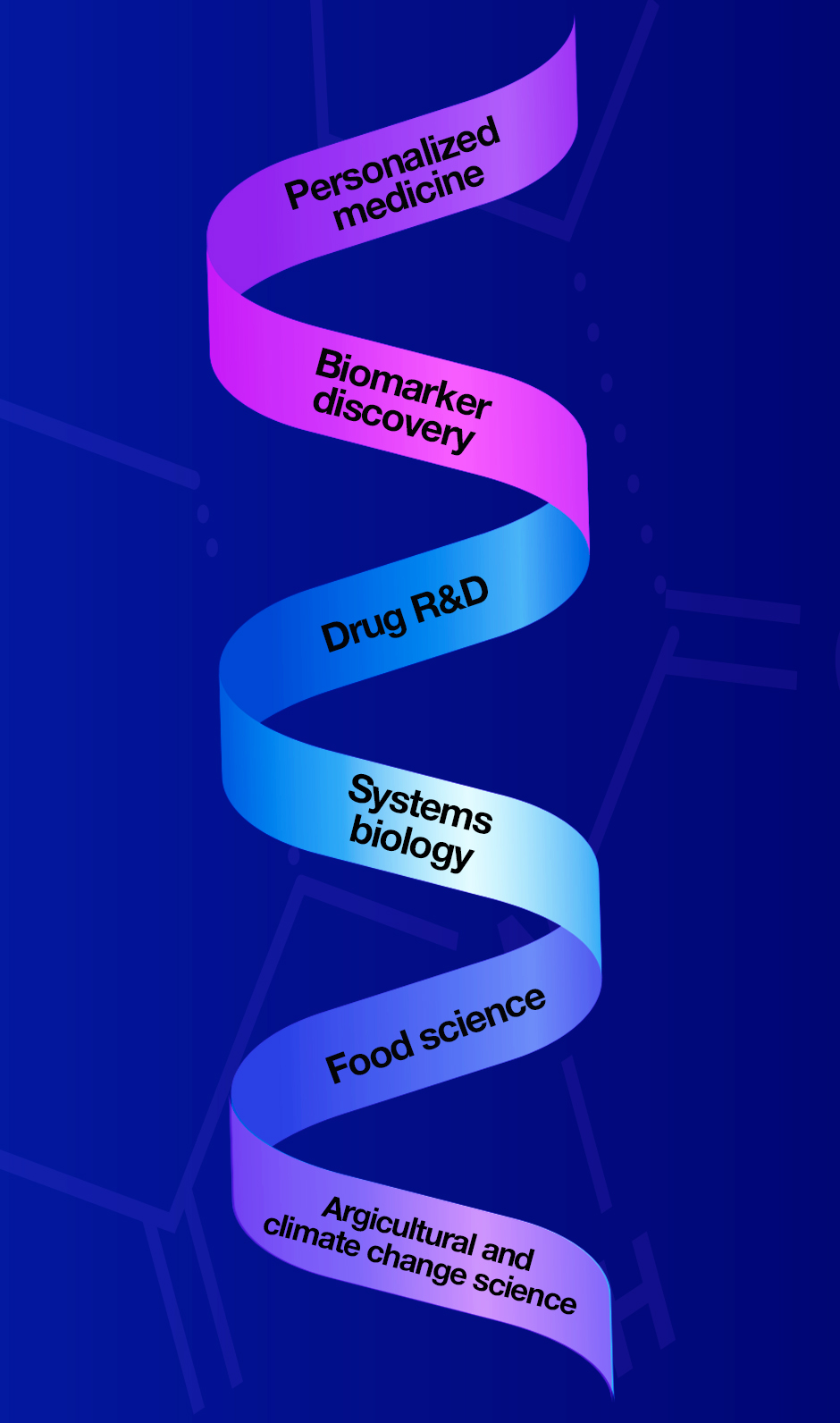
Personalized medicine: Placing the patient at the center of medicine, using their unique biological characteristics to tailor diagnostics and treatments.
Biomarker discovery: Identifying protein-based biomarkers that underpin specific states, such as a disease or a response to a treatment (e.g., how a drug is impacting a patient’s body at the molecular level).
Drug R&D: Identifying novel drug targets, developing new protein-based biopharmaceuticals.
Systems biology: Studying biological processes with a holistic approach, incorporating data from other omics principles, such as genomics and metabolomics.
Food science: Analyzing protein content of food, detection of allergens and supporting quality control assessments.
Agriculture and climate change science: Exploring interactions between plants, their environment and pathogens, for example. Engineering plants to increase their resilience to climate change-induced environmental changes.
“Everyone in proteomics stands on each other's shoulders […] One of the things that has been fantastic to watch is the way that so many people with different approaches have come together to try and make this all happen,”
– Professor Marc Wilkins said.