

Key Techniques in Structural Biology, Their Strengths and Limitations

Complete the form below to unlock access to ALL audio articles.
Structural biology is a field of science that uses a variety of techniques to determine the 3D structures of biomolecules such as proteins, nucleic acids and their complexes. These techniques allow researchers to elucidate the molecular architecture at different resolutions, from atomic to supramolecular levels. Additionally, structural biology focuses on the study of biomolecular interactions and dynamics, that is, how molecules interact and change over time. This is crucial for comprehending the function of biomolecules and their role in health and disease.
What techniques are used in structural biology?
Cryo EM
X ray crystallography
NMR spectroscopy
Cross-linking mass spectrometry (XL-MS)
Small-angle X-ray scattering (SAXS)
Neutron diffraction
Proteolysis
Circular dichroism (CD)
Electron paramagnetic resonance spectroscopy (EPR)
What techniques are used in structural biology?
The main techniques used in structural biology include X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy and cryogenic electron microscopy (cryo-EM), which are usually complemented with other methods, such as cross-linking mass spectrometry (XL-MS), small-angle X-ray scattering (SAXS), neutron diffraction, proteolysis, circular dichroism (CD) and electron paramagnetic resonance spectroscopy (EPR). Advances in computational methods and technologies have also played a significant role in structural biology, providing new ways to analyze, interpret and integrate data from different techniques. This has enabled researchers to gain a deeper understanding of biomolecules and their interactions, dynamics and relationship to biological processes. Let us consider these key techniques, their role in structural biology and their strengths and limitations.
Cryo EM
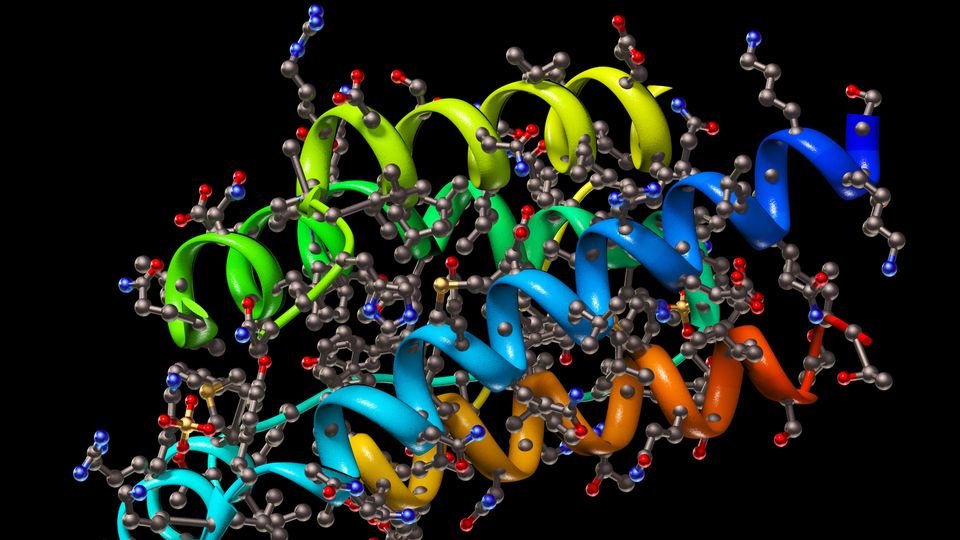
Cryo-EM is an electron microscopy technique in which the samples are subjected to a cryogenic treatment prior to analysis. Generally, it is applied to the structural study of proteins, especially those that cannot be studied by other techniques, such as large protein complexes, membrane proteins or proteins that do not form crystals. This technique allows images to be obtained with near-atomic resolution (comparable to X-ray crystallography or NMR spectroscopy) thanks to the interaction of an electron beam going through the sample. Cryogenizing the sample favors the preservation of the native molecular structures. By virtue of the recent developments of instrumentation and software, it is possible to process a very large number of images of the sample (for example, a protein) automatically and reconstruct its 3D structure with a near-atomic resolution (Figure 1).1, 2, 3, 4
Regarding its limitations, sample preparation in cryo-EM is arduous and some protein complexes can be destroyed during the process. Cryo-EM instruments and their maintenance are costly, and the huge amount of data generated requires a great computational effort to process it. Finally, there is a limitation in the size of the proteins that can be analyzed using cryo-EM because, below a certain size, the images obtained show very low signal-to-noise ratio.1, 2, 3, 4

Figure 1: Workflow of protein structure elucidation by cryo-EM. Credit: Technology Networks.
X ray crystallography
This technique is based on the fact that a crystalline sample is able to cause the diffraction of an incident X-ray beam, generating a characteristic pattern that can be used to deduce the structure of the crystal at an atomic level. The crystallized sample is irradiated with an X-ray beam at different angles of incidence. X-ray scattering caused by the ordered atomic structure of the sample generates characteristic patterns that are recorded by a sensor. The whole set of patterns obtained at different angles of incidence allows, by means of informatics processing, the electron density of the sample to be inferred from which it is possible to build a structural model at atomic scale (Figure 2).5, 6, 7, 8, 9
X-ray crystallography is one of the structural biology techniques with the highest level of resolution. It is well-established, with a high degree of automation and is relatively cheap compared to other structural biology approaches. Structure resolution processing is quite fast after data acquisition. Theoretically, there is no size limit to the molecules that can be analyzed by X-ray crystallography, but there are limitations imposed by the ease of sample crystallization. Large, complex molecules are often difficult to crystallize, especially when they contain dynamic areas, such as flexible regions or attached carbohydrates. Even when it is possible to generate a crystal of a complex molecule, sometimes it is difficult to elucidate their structure with a good resolution. Finally, the crystallization process can affect the molecular structure in a manner that means the final elucidated structure could be completely different to the native one found under physiological conditions.5, 6, 7, 8, 9, 10

Figure 2: Steps for protein structure determination by X-ray crystallography. Credit: Technology Networks.
As X-ray radiation is highly energetic, sample crystals used in X-ray crystallography must be flash-frozen with liquid nitrogen prior to measurement to minimize sample damage and reduce the noise produced by thermal motion. During the last decade, a variant of X-ray crystallography has been developed—serial femtosecond crystallography (SFX)—that negates this need. SFX is based in the use of an X-ray radiation source, called an X-ray free-electron laser (XFEL), that is able to generate very intense X-ray pulses of very short duration (in the order of fs = 10-15 s). These pulses are so powerful that they completely destroy the sample crystal. Therefore, it is necessary to carry out a serial analysis of a very high number of randomly oriented crystals (> 100,000) to elucidate the molecular structure of the sample. To achieve this, a liquid jet containing small crystals of the sample is injected into a chamber and irradiated by the XFEL. As pulses are extremely fast, thousands of crystals are analyzed in a very short time (Figure 3). The advantages of this approach are that crystals can be much smaller than those used in traditional X-ray crystallography and there is no need to flash-freeze them prior to measurement, therefore, experiments can be performed at room temperature. In addition, SFX is able to give information about molecular dynamics.11, 12, 13, 14, 15
 Figure 3: Experimental setup for SFX for protein structure determination. Credit: Liu&Lee, 2019,16 reproduced under the Creative Commons 4.0 International (CC BY 4.0) license. The electron source label has been updated for clarity with the article description.
Figure 3: Experimental setup for SFX for protein structure determination. Credit: Liu&Lee, 2019,16 reproduced under the Creative Commons 4.0 International (CC BY 4.0) license. The electron source label has been updated for clarity with the article description.
In the context of drug discovery, proteins are the main target for novel drug candidates. For this reason, it has been a major interest in the field of structural biology to elucidate the structure of more and more proteins. Advances in the procedures to obtain crystals rapidly from protein samples has led to the development of what is known as high-throughput (HT) crystallography. This approach is based on the use of automated systems, miniaturization and process integration in order to obtain large numbers of crystallized samples that can be analyzed in a short period of time, generating extensive sets of data in a very efficient way. This technique not only allows swift optimization of crystallization conditions, but is also useful to perform large-scale screenings for drug candidates that target proteins.17, 18, 19, 20, 21, 22
NMR spectroscopy
NMR spectroscopy takes advantage of a physical phenomenon in which the atomic nuclei are irradiated with radiofrequency and reach excited energy states. When nuclei recover their fundamental energy state, they emit a radiation that is collected by a detector (Figure 4). The frequency of the radiation detected depends on the chemical environment of the atomic nuclei and its intensity is related to the number of atomic nuclei of the same kind. There are multiple types of NMR experiments that provide information about different chemical elements (e.g., 1H, 2H, 13C and 15N), their connectivity or spatial proximity.23
The information given by NMR spectroscopy can be useful for many purposes, including:
- Structural elucidation of macromolecules
- Study of macromolecular dynamics
- Characterization of mechanism of interaction between biomolecules
For structural elucidation, the data obtained by NMR spectroscopy allows the distance between atoms and also bond angles to be calculated. These distance and angle values are used as restraints to perform computational calculations of the structure. In the case of molecular dynamics, some NMR experiments are used to analyze structural flexibility and estimate relaxation parameters. Finally, there are many experimental NMR approaches to detect interactions between molecules and identify the atoms involved, which enables the comprehension of interaction mechanisms, something very relevant in the drug discovery field.23, 24, 25, 26, 27
NMR spectroscopy is especially useful in the study of biomolecules under near-native conditions, as it is usually carried out in aqueous solution. It is a very versatile, non-destructive technique due to the large number of different experiments available oriented to analyze diverse molecular aspects. Together with cryo-EM and X-ray crystallography, NMR spectroscopy is one of the most used approaches for the structural determination of biomolecules.24, 25, 26, 27
However, NMR spectroscopy has its limitations. First, instruments and maintenance are quite expensive. Second, it has low sensitivity because most of the atomic nuclei that can be analyzed correspond to isotopes that show low natural occurrences. This means that many biomolecular samples must be enriched in some isotopes (such as 13C or 15N), a procedure that is costly. In addition, there is a limit to the size of the molecules that can be analyzed by solution NMR spectroscopy because large molecules can affect the homogeneity of the sample in aqueous solution, which is a critical point. In these cases, solid state NMR spectroscopy can be an alternative.23, 24, 25, 26, 27

Figure 4: NMR spectroscopy workflow for protein structural determination. Credit: Technology Networks.
Cross-linking mass spectrometry (XL-MS)
XL-MS is a type of mass spectrometry (MS) analysis in which a biological sample (isolated proteins, macromolecular complexes, organelles, cells or even tissues or organs) is treated with a cross-linking agent. Then, the sample is subjected to an enzymatic proteolysis and subsequently analyzed by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS).28, 29, 30, 31, ,32, 33, 34
The treatment with a cross-linking agent causes many attachments between different regions of proteins or other molecules that are interacting in the sample. When the sample is put through proteolysis, the proteins from the sample will fragment but the cross-linked regions will stay attached. Since the length of the cross-link is known, computational analysis of the resultant data will establish distance restraints between proteins and identify which ones are interacting. Taking all these data together, it is possible to describe protein interaction networks present in the sample (Figure 5).28, 29, 30, 31, 32, 33, 34
XL-MS is a highly sensitive technique capable of detecting and identify very low amounts of analyte (~10-15 mol). Contrary to other techniques, there is no limit in the size or complexity of the studied molecules, as the MS analysis is performed on the protein fragments. Since the cross-linking reaction can be conducted under physiological conditions, the results will reflect the native conditions. The major contribution of this technique is its great ability to generate high-throughput information about protein interaction networks, especially in challenging systems such as intrinsically disordered proteins (IDPs).28, 29, 30, 31, 32, 33, 34

It is very important to ensure high-quality MS data is obtained in this technique to minimize ambiguity in the assignment of the cross-linked species. The complete elucidation of isolated proteins or protein complexes is not possible by using XL-MS alone, it is necessary to use complementary techniques, such as cryo-EM.28, 29, 30, 31, 32, 33, 34
 experiment.")
Figure 5: General scheme of a typical cross-linking mass spectrometry (XL-MS) experiment. Credit: Technology Networks, adapted from Low et al., 2021.35
Small-angle X-ray scattering (SAXS)
SAXS is a technique based on the elastic scattering of X-ray photons after interaction with a sample, and collection of the radiation deviated at small angles (typically, 0.1-10°) with respect to the original beam trajectory.36, 37, 38, 39, 40
In a typical SAXS experiment, the sample is irradiated with X-rays and the radiation scattered at low angles is registered and represented as a curve of decaying intensity. The mathematical analysis of this curve enables the estimation of parameters related to the size and shape of the molecule. Using ab initio computational modelling on SAXS data, it is possible to build low-resolution molecular models that are very useful to determine the global shape of a macromolecule or the general disposition of structural domains within a protein (Figure 6). In this respect, these models can be combined with high-resolution structures of protein domains solved by other techniques (such as X-ray crystallography and NMR spectroscopy) to obtain a more detailed description of the molecular structure.36, 37, 38, 39, 40
SAXS is a useful tool for the study of non-crystalline biomolecular structures at low resolution. A wide range of molecular sizes (from kDa to GDa) and conditions (from native to extreme) can be analyzed with this technique. In addition, SAXS is capable of providing information about dynamics and kinetics of molecular processes. SAXS requires a small amount of sample, which generally is not destroyed during the measurement, and it can be prepared as liquid, solution or powder.36, 37, 38, 39, 40
The main limitation of SAXS is its lower resolution compared to other techniques, especially in solution or partially ordered samples. To gain resolution, it is necessary to employ powerful X-ray sources, such as synchrotrons, which are only available in large research facilities because of their cost and size. As proteins are labile molecules, some damage can occur during the measurements.36, 37, 38, 39, 40

Figure 6: Experimental scheme of a SAXS experiment to obtain structural information about a protein sample. Credit: Technology Networks.
Neutron diffraction
Neutron diffraction techniques are based on the irradiation of a sample with a neutron beam at different angles of incidence. The interaction of neutrons with the molecules of the sample causes their scattering, generating a characteristic pattern that depends on the sample structure at the atomic level (Figure 7). In this sense, the fundamentals are rather similar to those of X-ray diffraction techniques, but the main difference is the high penetrating capacity of neutrons compared to X-rays. For this reason, it is possible to obtain information about the sample bulk regardless its thickness. Neutrons are able to interact with atomic nuclei, rather than with their surrounding electron clouds (as X-rays do). As a consequence, neutron diffraction is sensitive to isotopic differences.6, 41, 42, 43, 44, 45
The samples analyzed by neutron diffraction must be in the form of a crystalline powder, although it is possible to use isolated crystals if they are large enough (larger than the ones required in X-ray crystallography). Scattering patterns obtained with this technique are analyzed to extract structural data about the molecule, similarly to X-ray crystallography (Figure 7). Nonetheless, one of the advantages of neutron diffraction with respect to X-ray diffraction is its capacity to provide information about hydrogen atoms. These atoms are almost invisible under X-ray radiation due to the fact that they have a very low electron density. Conversely, neutron diffraction is able to provide information about hydrogen bonds and the orientation of water molecules in the sample. Additionally, this technique can be applied to the study of dynamic properties of molecules, as it has a high spatial and time resolution.6, 41, 42, 43, 44, 45
Neutron diffraction is a technique showing some advantages compared to its X-ray counterpart, such as its ability to provide structural information involving light atoms, such as hydrogen and its capacity to discern between different isotopes. However, this technique presents some important drawbacks. First, neutron sources are very expensive to build and maintain. Second, for single-crystal analysis, the sample must be considerably larger than the ones used in X-ray crystallography. Finally, this technique may require long experimental times to acquire the data necessary to obtain high-resolution structural information.6, 41, 42, 43, 44, 45
 Scheme of a typical neutron diffraction experiment and the resulting spectrum. B) Neutron diffraction spectra fitting can be related to form and structure factors of the molecule analyzed.")
Figure 7: A) Scheme of a typical neutron diffraction experiment and the resulting spectrum. B) Neutron diffraction spectra fitting can be related to form and structure factors of the molecule analyzed. Credit: Castellanos et al., 2017,46 reproduced under the Creative Commons 4.0 International (CC BY 4.0) license.
Proteolysis
A useful approach to address protein structure and shape involves subjecting protein samples to a limited proteolysis prior to analysis. Proteolysis is the process in which proteins are broken down by enzymes into smaller peptides due to the hydrolysis of peptide bonds between amino acids. However, proteins in their native state are quite resistant to proteolysis and in many cases just one peptide bond is broken during the process. The reason behind this observation is that proteases are only capable of performing their function on protein regions with a certain degree of flexibility, such as loops, and thus have a limited activity. This can be explained by the fact that proteolysis occurring on peptide bonds located within secondary structure elements would require previous loss of numerous stabilizing interactions, making the process energetically unfavorable.47, 48, 49, 50, 51
There are many proteases that can be used to carry out proteolysis and they usually show amino acid specificity. After limited proteolysis, the resulting peptides can be isolated, analyzed and identified by other techniques (for instance, LC-MS) (Figure 8). In this context, a proteolysis approach can be used to identify flexible regions and structural domains in proteins, to study protein folding by generating folding intermediates and then conducting limited proteolysis on them and to characterize protein aggregation processes.47, 48, 49, 50, 51
Limited proteolysis has the advantage over other techniques of being applicable to samples in solution, that is, in native conditions. In addition, it is a simpler and cheaper approach compared to other physicochemical techniques, and it does not require any special instrumentation or large amounts of sample. However, limited proteolysis is not a high-resolution technique, and it is necessary to complement it with isolation and analysis procedures involving more sensitive techniques, such as LC-MS.47, 48, 49, 50, 51

Figure 8: Experimental workflow to study protein–drug interactions using a limited proteolysis approach. Credit: Technology Networks, adapted from Holfeld et al., 2023.52
Circular dichroism (CD)
CD spectroscopy is a technique based on the interaction between circularly polarized light and matter. Electromagnetic radiation is composed of an electric field oscillating perpendicularly to a magnetic field (both in the xy plane). The plane containing both fields is, in turn, perpendicular to the direction in which the wave propagates (z axis). When radiation is polarized, its electric field oscillates with a specific geometric orientation. In the case of circular polarization, the electric field of the radiation rotates at a constant rate describing a circumference. This rotation can be clockwise (right circularly polarized light, RCP) or counterclockwise (left circularly polarized light, LCP). Circularly polarized radiation is able to interact with optically active molecules (chiral molecules); these are molecules containing one or more atoms that are bound to four different substituents.53, 54, 55, 56, 57
In a typical CD spectroscopy experiment, the sample is irradiated with a range of circularly polarized light of different wavelengths, typically from the ultraviolet to visible range. As proteins are optically active molecules, they are able to absorb part of the radiation. RCP and LCP are differently absorbed by the sample and this difference (Δε) is called “circular dichroism” (Figure 9A). Experimental results are displayed representing Δε as a function of the wavelength. The shape of the resultant curve can be related with the secondary structure of the protein (Figure 9B) and in some cases it is possible to obtain a quantitative estimation of the content of some secondary structure elements in the sample.53, 54, 55, 56, 57
CD spectroscopy is a simple and relatively inexpensive technique that allows information to be gained about the secondary structure and folding/unfolding of proteins and peptides. CD experiments are easy to perform and not very time-consuming. Proteins and peptides can be studied in native conditions, as measurements are carried out in solution and only low concentrations of the analyte are required (~μM).53, 54, 55, 56, 57
Some of the limitations of this technique include the fact that some chemical species can interfere in the measurements (substances that absorb at the same wavelengths, such as many buffers and chloride). It is difficult to deduce detailed structural information of complex molecules, and quantification of secondary structure elements is only reliable when analyzing small peptides.53, 54, 55, 56, 57
 Scheme of a circular dichroism experiment indicating all the elements of the CD spectrometer. B) Examples of the typical CD spectra of different elements of secondary structure in proteins.")
Figure 9: A) Scheme of a circular dichroism experiment indicating all the elements of the CD spectrometer. B) Examples of the typical CD spectra of different elements of secondary structure in proteins. Credit: Pignataro et al., 2020,58 reproduced under the Creative Commons 4.0 International (CC BY 4.0) license.
Electron paramagnetic resonance spectroscopy (EPR)
EPR spectroscopy is based on the interaction between microwave radiation and the electronic spin of unpaired electrons. The working principle is very similar to that of NMR spectroscopy: the sample is placed within an external magnetic field and then irradiated with microwave radiation (radiofrequency in NMR) that is absorbed by unpaired electrons (atomic nuclei in NMR), reaching excited states. When the unpaired electrons recover their fundamental energy state, the sample emits radiation that is detected and processed to generate an EPR spectrum, from which various types of structural information can be deduced (Figure 10).59, 60, 61, 62, 63, 64
Biological molecules are not usually paramagnetic, meaning that they do not contain unpaired electrons (paramagnetic molecules, like radicals, are very unstable). Therefore, it is necessary to transform them into paramagnetic molecules prior to the EPR analysis. The most common way to do this is by using spin-labeling methods, which consist of attaching a non-reactive paramagnetic chemical group (typically nitrogen-containing groups) to the biomolecule. In the case of proteins, site-directed spin-labeling (SDSL) is used to introduce unpaired electrons in specific amino acids. EPR can be used to investigate many aspects of biological molecules, such as molecular motion and dynamics, and the presence of different secondary structure elements. Through this technique, it is also possible to obtain measurements of distance between paramagnetic centers within the molecule. Those distances can be used as restraints to build molecular models.59, 60, 61, 62, 63, 64
EPR spectroscopy offers a high spatial (nm) and temporal (ps–μs) sensitivity, which is very useful to study protein dynamics, especially in highly dynamic proteins (such as IDPs). Despite the fact that it is necessary to use spin labels in the analyzed molecules, their small size generates minimal perturbations in the system. There is no size limit to the molecule analyzed and spectra are simpler than in other techniques as only unpaired electrons are visible. This technique provides relevant structural information, but it should be complemented with other high-resolution techniques to get a complete structural model of the analyzed biomolecule. In addition, spin electrons couple with nearby nuclear spins resulting in signal splitting and broadening, which reduces resolution.59, 60, 61, 62, 63, 64
 Scheme of a typical EPR spectrometer. B) EPR spectra from an experiment oriented to the study of protein folding in the presence and the absence of a protein activator.")
Figure 10: A) Scheme of a typical EPR spectrometer. B) EPR spectra from an experiment oriented to the study of protein folding in the presence and the absence of a protein activator. These spectra reflect the alterations in protein folding between the native and the misfolded states, and between the active and inactive form in the correctly folded protein. Credit: Technology Networks, adapted from Balo et al., 2019.65
To find out more about structural biology, what it can tell us and its applications, visit the article below.

References
1. Earl LA, Falconieri V, Milne JLS, Subramaniam S. Cryo-EM: Beyond the microscope. Curr Opin Struc Biol. 2017; 46:71-78. doi:10.1016/j.sbi.2017.06.002
2. Nogales E, Scheres SHW. Cryo-EM: A unique tool for the visualization of macromolecular complexity. Mol Cell. 2015. 58(4):677-689. doi:10.1016/j.molcel.2015.02.019
3. Bai X-C, McMullan G, Scheres SHW. How cryo-EM is revolutionizing structural biology. Trends Biochem Sci. 2015; 40(1):49-57. doi:10.1016/j.tibs.2014.10.005
4. Egelman, EH. The current revolution in cryo-EM. Biophys J. 2016; 110:1008-1012. doi:10.1016/j.bpj.2016.02.001
5. Ameh ES. A review of basic crystallography and x-ray diffraction applications. Int J Adv Manufac Tech. 2019; 105:3289-3302. doi:10.1007/s00170-019-04508-1
6. Egli M. Diffraction techniques in structural biology. Curr Protoc Nucleic Acid Chem. 2017; 65:7.13.1-7.13.41. doi:10.1002/cpnc.4
7. Maveyraud L, Mourey L. Protein X-ray crystallography and drug discovery. Molecules. 2020; 25(5):1030. doi:10.3390/molecules25051030
8. Muench SP, Antonyuk SV, Hasnain SS. The expanding toolkit for structural biology: Synchrotrons, X-ray lasers and cryo-EM. IUCrJ. 2019; 6:167-177. doi:10.1107/S2052252519002422
9. Shi Y. A glimpse of structural biology through X-ray crystallography. Cell. 2014; 159(5):995-1014. doi:10.1016/j.cell.2014.10.051
10. Acharya KR, Lloyd MD. The advantages and limitations of protein crystal structures. Trends Pharmacol Sci. 2005; 26(1):10-14. doi:10.1016/j.tips.2004.10.011
11. Johansson LC, Stauch B, Ishchenko A, Cherezov V. A bright future for serial femtosecond crystallography with XFELs. Trends Biochem Sci. 2017. 42(9):749-762. doi:10.1016/j.tibs.2017.06.007
12. Martin-Garcia JM, Conrad CE, Coe J, Roy-Chowdhury S, Fromme P. Serial femtosecond crystallography: A revolution in structural biology. Arch Biochem Biophys. 2016; 602:32-47. doi:10.1016/j.abb.2016.03.036
13. Orville AM. Recent results in time resolved serial femtosecond crystallography at XEFLs. Curr Opin Struc Biol. 2020; 66:193-208. doi:10.1016/j.sbi.2020.08.011
14. Schlichting I. Serial femtosecond crystallography: The first five years. IUCrJ. 2015; 2(2):246-255. doi:10.1107/S205225251402702X
15. Schriber EA, Paley DW, Bolotovsky R., Rosenberg DJ, Sierra RG, Aquila A, et al. Chemical crystallography by serial femtosecond X-ray diffraction. Nature. 2022; 601(7893):360-365. doi:10.1038/s41586-021-04218-3
16. Liu H & Lee W. Protein crystallography: Developments and perspectives. Int J Mol Sci. 2019; 20(14):3421. doi:10.3390/ijms20143421
17. Tickle I, Sharff A, Vinković M, Yon J, Jhoti H. High-throughput protein crystallography and drug discovery. Chem Soc Rev. 2004; 33:558-565. doi:10.1039/B314510G
18. Sutanto F, Shaabani S, Oerlemans R, Eris D, Patil P, Hadian M, et al. Combining high-throughput synthesis and high-throughput protein crystallography for accelerated hit identification. Angew Chem Int Ed. 2021; 60:18231-18239. doi:10.1002/anie.202105584
19. Stevens RC. High-throughput protein crystallization. Curr Opin Struc Biol. 2000; 10(5):558-563. doi:10.1016/S0959-440X(00)00131-7
20. Sharff A, Jhoti H. High-throughput crystallography to enhance drug discovery. Curr Opin Chem Biol. 2003; 7(3):340-345. doi:10.1016/S1367-5931(03)00062-0
21. Rupp B. High-throughput crystallography at an affordable cost: The TB structural genomics consortium crystallization facility. Acc Chem Res. 2003; 36(3):173-181. doi:10.1021/ar020021t
22. Goodwill KE, Tennant MG, Stevens RC. High-throughput X-ray crystallography for structure-based drug desing. Drug Discov Today. 2001; 6(15):S113-S118. doi:10.1016/S1359-6446(01)00169-6
23. Wüthrich K. Protein structure determination in solution by NMR spectroscopy. J Biol Chem. 1990; 265(36):22059-22062. doi:10.1016/S0021-9258(18)45665-7
24. Sugiki T, Furuita K, Fujiwara T, Kojima C. Current NMR techniques for structure-based drug discovery. Molecules. 23(148):1-27. doi:10.3390/molecules23010148
25. Opella SJ, Marassi FM. Applications of NMR to membrane proteins. Arch Biochem Biophys. 2017; 628:92-101. doi:10.1016/j.abb.2017.05.011
26. Markwick PRL, Malliavin T, Nilges M. Structural biology by NMR: Structure, dynamics, and interactions. PLoS Comput Biol. 2008; 4(9):e1000168. doi:10.1371/journal.pcbi.1000168
27. Van der Wel, PCA. New applications of solid-state NMR in structural biology. Emerg Top Life Sci. 2018; 2:57-67. doi:10.1042/ETLS20170088
28. Iacobucci C, Sinz A. To be or not to be? Five guidelines to avoid misassignments in cross-linking/mass spectrometry. Anal Chem. 2017; 89:7832-7835. doi:10.1021/acs.analchem.7b02316
29. Iacobucci C, Götze M, Sinz A. Cross-linking/mass spectrometry to get a closer view on protein interaction networks. Curr Opin Biotech. 2020; 63:48-53. doi:10.1016/j.copbio.2019.12.009
30. Fischer L, Chen ZA, Rappsilber J. Quantitative cross-linking/mass spectrometry using isotope-labelled cross-linkers. J Proteomics. 2013; 88:120-128. doi:10.1016/j.jprot.2013.03.005
31. Piersimoni L, Sinz A. Cross-linking/mass spectrometry at the crossroads. Anal Bioanal Chem. 2020; 412:5981-5987. doi:10.1007/s00216-020-02700-x
32. Sinz A. Cross-linking/mass spectrometry for studying protein structures and protein-protein interactions: Where are we now and where should we go from here? Angew Chem Int Ed. 2018; 57:6390-6396. doi:10.1002/anie.201709559
33. Yu C, Huang L. Cross-linking mass spectrometry (XL-MS): An emerging technology for interactomics and structural biology. Anal Chem. 2018; 90(1):144-165. doi:10.1021/acs.analchem.7b04431
34. Tang X, Wippel HH, Chavez JD, Bruce JE. Crosslinking mass spectrometry: A link between structural biology and systems biology. Protein Sci. 2021; 30:773-784. doi:10.1002/pro.4045
35. Low TY, Syafruddin SE, Mohtar MA, Vellaichamy A, Rahman NSA, Pung Y-F, Tan CSH. Recent progress in mass spectrometry-based strategies for elucidating protein-protein interactions. Cell Mol Life Sci. 2021; 78:5325-5339. doi:10.1007/s00018-021-03856-0
36. Doniach S. Changes in biomolecular conformation seen by small angle X-ray scattering. Chem Rev. 2001; 101:1763-1778. doi:10.1021/cr990071k
37. Gräwert TW, Svergun DI. Structural modeling using solution small-angle X-ray scattering (SAXS). J Mol Biol. 2020; 432:3078-3092. doi:10.1016/j.jmb.2020.01.030
38. Lipfert J, Doniach S. Small-angle X-ray scattering from RNA, proteins, and protein complexes. Annu Rev Biophys Struct. 2007; 36:307-327. doi:10.1146/annurev.biophys.36.040306.132655
39. Petoukhov MV, Svergun DI. Applications of small-angle X-ray scattering to biomacromolecular solutions. Int J Biochem Cell B. 2013; 45(2):429-437. doi:10.1016/j.biocel.2012.10.017
40. Kikhney AG, Svergun DI. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Let. 589(19):2570-2577. doi:10.1016/j.febslet.2015.08.027
41. Blasie JK, Timmins P. Neutron scattering in structural biology and biomolecular materials. MRS Bull. 24(12):40-47. doi:10.1557/S0883769400053719
42. Blakeley MP, Langan P, Niimura N, Podjarny A. Neutron crystallography: Opportunities, challenges, and limitations. Curr Opin Struc Biol. 2008; 18:593-600. doi:10.1016/j.sbi.2008.06.009
43. Hoogerheide DP, Forsyth VT, Brown KA. Neutron scattering for structural biology. Phys Today. 2020; 73(6):36-42. doi:10.1063/PT.3.4498
44. Kono F, Kurihara K, Tamada T. Current status of neutron crystallography in structural biology. BPPB. 2022; 19:e190009. doi:10.2142/biophysico.bppb-v19.0009
45. Niimura N. Neutrons expand the field of structural biology. Curr Opin Struc Biol. 1999; 9(5):602-608. doi:10.1016/S0959-440X(99)00012-3
46. Castellanos MM, McAuley A, Curtis JE. Investigating structure and dynamics of proteins in amorphous phases using neutron scattering. Comput Struct Biotech J. 2017; 15:117-130. doi:10.1016/j.csbj.2016.12.004
47. Duan Y, Liu Y, Coreas R, Zhong W. Mapping molecular structure of protein locating on nanoparticles with limited proteolysis. Anal Chem. 2019; 91:4204-4212. doi:10.1021/acs.analchem.9b00482
48. Fontana A, Laureto PP, Spolaore B, Frare E, Picotti P, Zambonin M. Probing protein structure by limited proteolysis. Acta Biochim Pol. 2004; 51(2):299-321. doi:10.18388/abp.2004.3573
49. Fontana A, Laureto PP, Spolaore B, Frare E. Identifying disordered regions in proteins by limited proteolysis. In: Uversky V, Dunker A, eds. Intrinsically Disordered Protein Analysis. Methods in Molecular Biology. New York: Springer; 2012:297-318. doi:10.1007/978-1-4614-3704-8_20
50. Fontana A, Laureto PP, Filippis V, Scaramella E, Zambonin M. Probing the partly folded states of proteins by limited proteolysis. 1997; 2(2):R17-R26. doi:10.1016/S1359-0278(97)00010-2
51. Kazanov MD, Igarashi Y, Eroshkin AM, Cieplak P, Ratnikov B, Zhang Y, Li Z, Godzik A, Osterman AL, Smit JW. Structural determinants of limited proteolysis. J Proteome Res. 2011; 10:3642-3651. doi:10.1021/pr200271w
52. Holfeld A, Quast JP, Bruderer R, Reiter L, de Souza N, Picotti P. Limited proteolysis-mass spectrometry to identify metabolite-protein interactions. In: Skirycz A, Luzarowski M, Ewald JC (eds). Cell-wide identification of metabolite-protein interactions. Meth Mol Biol. 2023; 2554:69-89. doi:10.1007/978-1-0716-2624-5_6
53. Kelly SM, Price NC. The use of circular dichroism in the investigation of protein structure and function. Curr Protein Pept Sc. 2000; 1(4):349-384. doi:10.2174/1389203003381315
54. Kelly SM, Jess TJ, Price NC. How to study proteins by circular dichroism. Biochim Biophys Acta. 2005; 1751(2):119-139. doi:10.1016/j.bbapap.2005.06.005
55. Martin SR, Schilstra MJ. Circular dichroism and its application to the study of biomolecules. Method Cell Biol. 2008; 84:263-293. doi:10.1016/S0091-679X(07)84010-6
56. Ranjbar B, Gill P. Circular dichroism techniques: Biomolecular and nanostrucural analyses – A review. Chem Biol Drug Des. 2009; 74:101-120. doi:10.1111/j.1747-0285.2009.00847.x
57. Whitmore L, Wallace BA. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers. 89(5):392-400. doi:10.1002/bip.20853
58. Pignataro MF, Herrera MG, Dodero VI. Evaluation of peptide/protein self-assembly and aggregation by spectroscopic methods. Molecules. 2020; 25(20):4854. doi:10.3390/molecules25204854
59. Biswas R, Kühne H, Brudvig GW, Gopalan V. Use of EPR spectroscopy to study macromolecular structure and function. Sci Prog. 2001; 84(1):45-68. doi:10.3184/003685001783239050
60. Drescher M. EPR in protein science. In: Drescher M, Jeschke G, eds. EPR Spectroscopy. Berlin: Springer. 2011: 91-119. doi:10.1007/128_2011_235
61. Jeschke G. The contribution of modern EPR to structural biology. Emerg Top Life Sci. 2018; 2:9-18. doi:10.1042/ETLS20170143
62. Li H, Pan Y, Yang Z, Rao J, ChenB. Emerging applications of site-directed spin labeling electron paramagnetic resonance (SDSL-EPR) to study food protein structure, dynamics, and interaction. Trends Food Sci Tech. 2021; 109:37-50. doi:10.1016/j.tifs.2021.01.022
63. Sahu ID, McCarrick RM, Lorigan GA. Use of electron paramagnetic resonance to solve biochemical problems. Biochemistry. 2013; 52:5967-5984. doi:10.1021/bi400834a
64. Sahu ID, Lorigan GA. Electron paramagnetic resonance as a tool for studying membrane proteins. Biomolecules. 2020; 10(5):763. doi:10.3390/biom10050763
65. Balo AR, Lee J, Ernst OP. Stationary phase EPR spectroscopy for monitoring membrane protein refolding by conformational response. Anal Chem. 2019; 91:1071-1079. doi:10.1021/acs.analchem.8b04542

