
Written by
Sponsored by

T
he pharmaceutical industry primarily strives to bring new medicines to the market via a complex multi-step process that involves the “discovery” and “development” of a drug compound. Bringing a new drug from “bench to bedside” can take up to 15 years and is a costly endeavor – to develop a small molecule compound or biotherapeutic it is estimated to cost, on average, $2.6 billion. Before a drug is approved for use by the relevant regulatory authorities, it must go through rigorous testing to confirm its efficacy and safety, and several key considerations will be evaluated such as the most appropriate administration route for the drug, manufacturing suitability, cost-effectiveness and commercial viability.
In this article, we take a closer look at the various stages of drug development and highlight the instrumental role drug discovery plays in the process.
Click on the individual drug development steps to learn more.


Drug discovery is the process whereby therapeutic targets and novel compounds with therapeutic potential are discovered. It involves multiple disciplines including biology, chemistry, pharmacology and bioinformatics. Once a lead compound is identified it undergoes further optimization before progressing to preclinical testing.
Preclinical studies are performed prior to clinical trials and are designed to assess a drug candidate’s efficacy and safety before it can be administered to a human. These studies can be carried out using in vivo or in vitro model systems. Choosing an appropriate preclinical model is extremely important to ensure reliable results regarding biological effect, dosing and toxicity.
The clinical development of an investigational new drug follows a series of phases. It is at this stage that the drug is tested in human subjects. Clinical trials must follow a research protocol. This document outlines the study’s rationale, key objectives, design, methodology and statistical factors.
An investigational new drug (IND) application must be submitted to the regulatory agency to request permission to administer an investigational medicine to humans. The application must contain information on the followingfollowing: preclinical studies, manufacturing, proposed clinical research protocols and clinical investigators.
The regulatory authority is responsible for the scientific evaluation of the new drug application (NDA)/marketing authorisation application (MAA).
Drug sponsors must submit an NDA to the regulatory authority before a new medicine can be sold and marketed. The application should contain comprehensive information about the drug to enable a reviewer to make an informed decision as to whether or not it should be approved.
Once the drug product receives approval from the regulatory authority, numerous activities are initiated to prepare for product launch, including manufacturing scale-up. The FDA provides guidance on “advanced manufacturing” which is designed to improve drug quality, address shortages of medicines and expedite time-to-market.
Post-marketing safety surveillance involves monitoring a drug product’s long-term safety and efficacy post-approval, when it has reached the market and is in wider use.
A
drug discovery project is typically initiated to address an unmet clinical need for an effective therapeutic for a particular indication. The process begins with initial or “basic” research, which is often conducted by academics. The primary goal at this stage is to identify a protein or pathway implicated in a disease or condition of interest, with the potential to be therapeutically targeted. It can take a number of years to gather enough supporting evidence before a target is selected for a drug discovery project – which is estimated to cost > $600 million. Once a target has been selected, focus shifts towards identifying molecules with suitable characteristics to make a drug. There are several subsequent steps involved and various approaches can be adopted.
Click on the individual drug discovery steps to find out more.
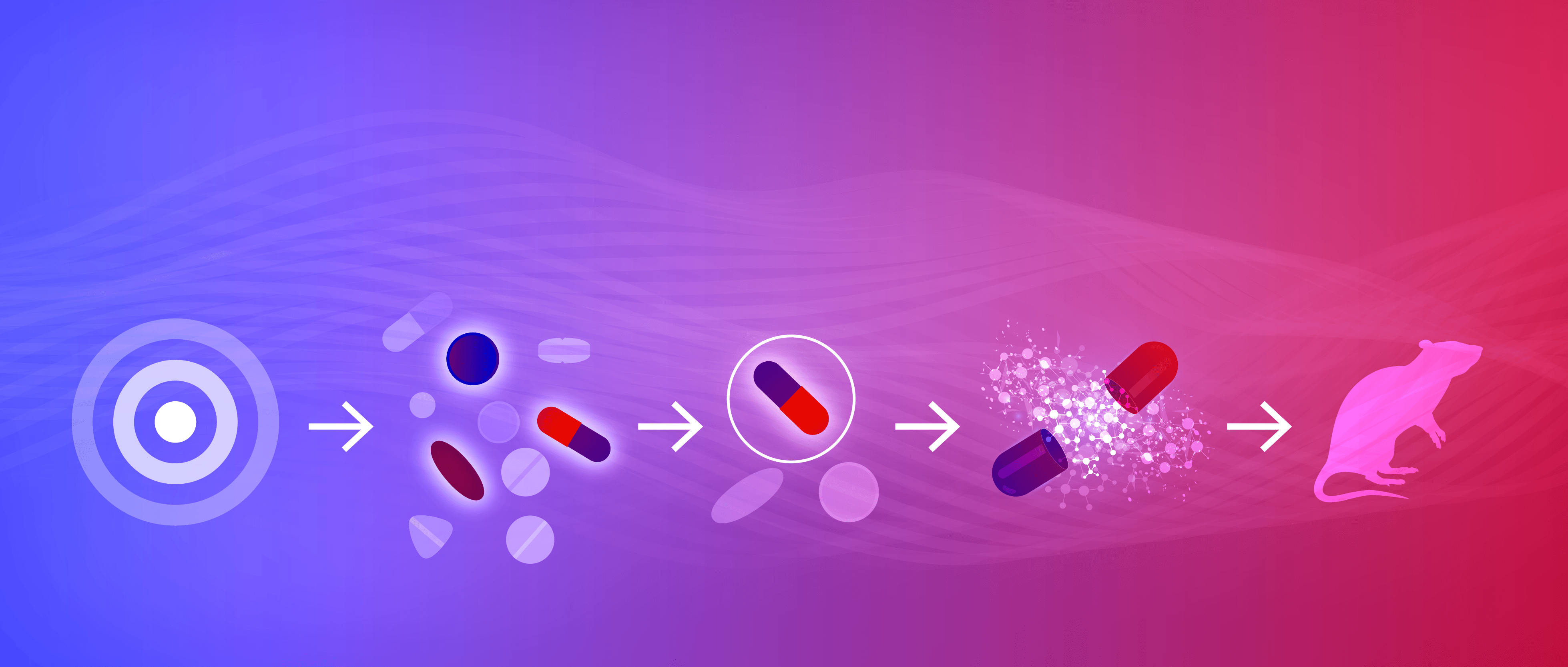
Once a target is identified it is validated to confirm its suitability for pharmaceutical development.
Screening approaches are exploited to identify “hits” – compounds that interact with the target of interest.
The most promising “hits” are refined to produce “leads” with improved properties.
Lead compounds are further optimized.
A drug candidate is selected for preclinical testing using in vitro and in vivo models.
1. Target identification and validation
Understanding the clinical spectrum of a disease and the role of a potential biological target in disease pathology is key to developing an efficacious drug. Targets come in various forms – receptors, enzymes, proteins, genes and RNA. For a target to be considered “druggable” its activity must be able to be modulated by a therapeutic agent. Certain targets are more amendable to small molecule discovery (e.g., G protein-coupled receptors) whereas others are modulated more effectively by biotherapeutics (e.g., protein antigen [target] and antibodies [biotherapeutic]).
Potential drug targets can be identified in different ways – by reviewing published literature, patent records and scouring open-access databases (data mining), or via experimental methods in the lab.
Broadly, target identification strategies can be split into two main categories: target deconvolution and target discovery (Figure 1). Target deconvolution involves the retrospective identification of a drug target in response to an observed (desirable) phenotypic response (from conducting phenotypic screening). Various techniques can be employed for target deconvolution including affinity chromatography, expression cloning, protein microarrays and biochemical suppression. Target discovery works on the premise that you want a drug compound and therefore a target must be identified to enable hit identification. The known target is then used to design relevant systems-based assays.
2. Hit identification and validation
Numerous screening approaches can be used to identify a “hit” compound. Hit is the term used to describe a compound that interacts with the target of interest. The output from these approaches provides the basis upon which drug design and elaboration are used to generate lead compounds with desirable properties.
High-throughput screening (HTS) involves the generation of a large library of compounds. These assays can be used to screen various library types (e.g., combinatorial chemistry, genomics, protein and peptide); thousands to millions of compounds are screened in parallel against a target. This screening approach is fast, cheap and relies heavily on automation. The success of HTS has been heavily influenced by advances in liquid-handing robots and miniaturization.
“The most fruitful basis for the discovery of a new drug is to start with an old drug,”
– Sir James W. Black, recipient of the 1988 Nobel Prize in Physiology or Medicine.
Virtual screening (VS) exploits computational approaches to “virtually screen” compounds to identify potential hits. Typically, large databases of commercially available compounds are virtually screened, however libraries of structures generated in silico from existing ligands can also be utilized to identify novel hits. VS can be a far more cost-effective approach to identify initial hits, as it circumvents the need to “physically screen” vast libraries against a biological target.
Fragment-based screening utilizes biophysical approaches to determine the binding of small “fragments” to a target. Initial fragment hits typically have a weak binding affinity to the target and associate with the target via “hot spots”. Fragment screening methods include NMR spectroscopy, SPR spectroscopy, X-ray crystallography, microscale thermophoresis, thermal shift assay and weak affinity chromatography. The initial fragments hits are subsequently expanded to produce larger molecules with higher binding affinity and are optimized to achieve a more desirable pharmacokinetic profile.
Identified hits are ranked and clustered according to their performance in several follow up experiments. Hits may be grouped based on structural similarity, to ensure various chemical classes are being considered. Dose-response curves are generated to compare potencies and in vitro assays are conducted to generate data on the absorption, distribution, metabolism and excretion (ADME) of the drug.
3. Hit-to-lead and lead optimization
Once the most promising hits have been confirmed, the next step is to refine them to produce compounds with higher potency and better selectivity. This step reduces the chance of off-target interactions which can lead to adverse effects. Medicinal chemists will work to increase the affinity of the compounds to the target of interest by several orders of magnitude. ADME properties are explored in more depth at this stage and the compounds are tested using in vivo disease models to determine pharmacokinetic profiles. Solubility and permeability assessments are carried out to determine the best route of administration and to eliminate compounds that lack the required properties to become a viable drug.
The goal of lead optimization is to retain advantageous properties previously defined, while optimizing the structure of each compound.
4. Candidate selection and preclinical testing
At this stage you will need to determine, from several promising leads, which one you want to take forward as a drug candidate for preclinical and clinical testing. Medicinal chemists will likely continue to produce “back up” molecules in case the lead selected for preclinical and clinical development fails. All the information gathered about the chosen molecule up until this point is used to construct a “target candidate profile” which is used as part of the investigational new drug (IND) application, for submission to the regulatory authorities.
